第4章 热力学
熵和玻耳兹曼的有序性原理
本书第2章和第3章所讨论的是和可逆现象相对应的时间的物理学,因为无论是哈密顿方程还是薛定谔方程,对于t→-t的替换都是不变的。这种情况我称之为存在的物理学。现在我们转到演化的物理学,说得明确一点,转到热力学第二定律所描述的不可逆过程。在本章和随后的两章中,我们将严格地采取唯象的观点。我们将不问可能与动力学有什么关系,但我们要概括出一些方法,这些方法在很宽的范围(从热传导这样的简单不可逆过程直到包含自组织的复杂过程的范围)内,成功地描述了单向的时间现象。
从热力学第二定律的表述开始,它就强调了不可逆过程所起的独特的作用。汤姆孙(William Thomson,又称Lord Kelvin,即开尔文勋爵)首次给出第二定律一般表述的论文题就是:“论自然界中机械能耗散的普遍倾向”(Thomson,1852)。克劳修斯还使用了宇宙学的语言:“宇宙的熵趋于最大”(Clausius,1865)。不过,必须承认,第二定律的表述对于我们今天来说,更像是一个纲领而不像是十分确定的陈述。因为无论汤姆孙还是克劳修斯,都没有给出任何方法,用可观察量来表示熵的变化。这个表述之缺乏明确性,大概就是为什么热力学的应用很快就局限于平衡态即热力学进化终态的原因之一。例如,吉布斯的在热力学史上有过如此影响的经典著作就曾小心地回避了对非平衡过程这个领域的任何干预(Gibbs,1875)。另一个原因很可能就是,在许多问题中不可逆过程是个很讨厌的东西,例如,它是获得热机最大效率的一个障碍。因此制造热机的工程师们的目标,就是使不可逆过程所带来的损失达到极小。
只是到了最近,才出现了看法上的彻底改变,而且我们开始理解了不可逆过程在物质世界中所起的建设性作用。当然,尽管如此,平衡态的情况仍然是最简单的。因为在这种情况下,与熵有关的变量的数目最少。让我们先简要地回顾一下某些经典的论点。
我们考虑一个只和外界交换能量,不和外界交换物质的系统,这样的系统称为封闭系统。与此相反,和外界既交换能量也交换物质的系统叫作开放系统。假设这个封闭系统处于平衡态,因此熵产生为零。另一方面,宏观熵的改变由从外界所得到的热量来决定。按定义,

其中T是个正的量,称做绝对温度。
让我们把这个关系式与适合这种简单系统的热力学第一定律结合起来(详见Prigogine,1967):

其中E是能量,p是压强,V是体积。这个公式表明,在一个小的时间间隔dt内,系统与外界交换的能量等于系统所获得的热量加上在其边界上所作的机械功。合并方程4.1和方程4.2,我们得到用变量E和V表达的熵的全微分

吉布斯把这个公式推广了,使它包括组分的改变量。令n1 ,n2 ,n3 ,…是各组分的克分子数,于是我们可以写出
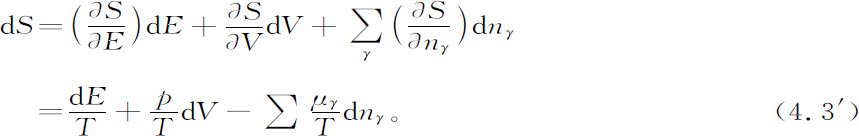
量μγ 按定义是吉布斯所引入的化学势,方程4.3′称做熵的吉布斯公式。化学势本身是热力学变量如温度、压强、浓度等的函数。对于所谓理想系统 (1) 化学势取特别简单的形式,即化学势与组分的克分子数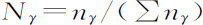 的对数有关系:
的对数有关系:

其中R是气体常数(等于玻耳兹曼常数k与阿伏伽德罗数的乘积),ζγ (p,T)是压强和温度的某个函数。
除了熵以外,人们常引入其他的热力学势,比如亥姆霍兹自由能,其定义如下:

很容易证明,适用于孤立系统的熵增加定律可以适用于恒温系统的自由能减少定律来代替。
方程4.5的结构反映了能量E和熵S之间的竞争。我们知道,在低温下第二项可以忽略,F的最小值给出相应于最小能量而且一般也相应于低熵的结构。但随着温度的增加,系统就变到熵越来越高的结构。
经验证明了这些想法,因为在低温下我们看到以低熵有序结构为特点的固态,而在高温时我们看到高熵的气态。物理学中一定类型的有序结构的形成乃是热力学定律应用于热平衡封闭系统的一个结果。
在第1章中我们曾给出玻耳兹曼用配容对熵所作的简单解释。让我们把这个公式用于能级为E1 ,E2 ,E3 的系统。在总能量和粒子数固定时,通过寻找使配容数(式1.9)为最大的占据数,我们得到玻耳兹曼基本公式,以求出占据给定能级Ei 的概率Pi :

其中k和式1.10中一样,是玻耳兹曼常数,T是温度,Ei 是所选能级的能量。假设我们考虑一个仅有三个能级的简化系统,那么玻耳兹曼公式即方程4.6将告诉我们在平衡时,这三个态中的每一个态中找到分子的概率。在极低的温度下,即T→0,只有对应于最低能级的概率是重要的,因此我们得到图4.1的情形,其中所有分子实际上都处在最低的能级E1 ,因为

但在高温下,三个概率将变得大体上相等,即

因此这三个态被近似均等地占据(见图4.2)。
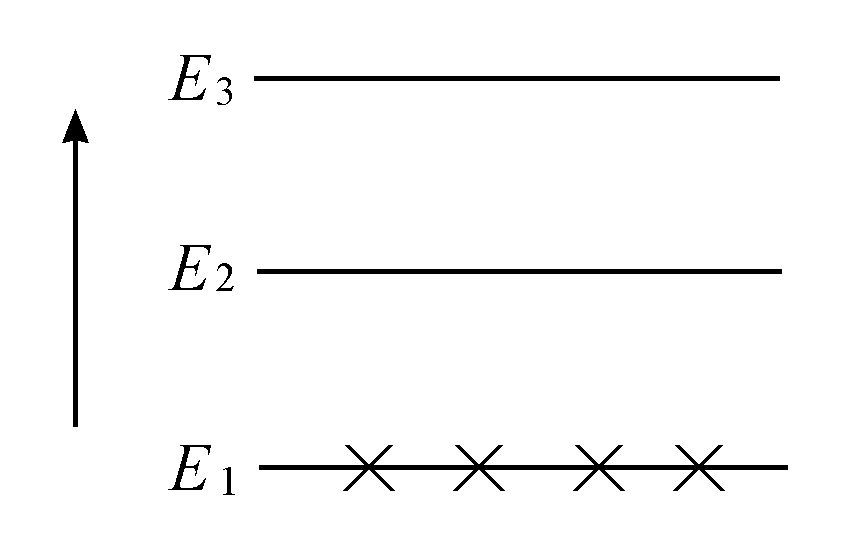
图4.1 低温分布
只有最低能级被占据
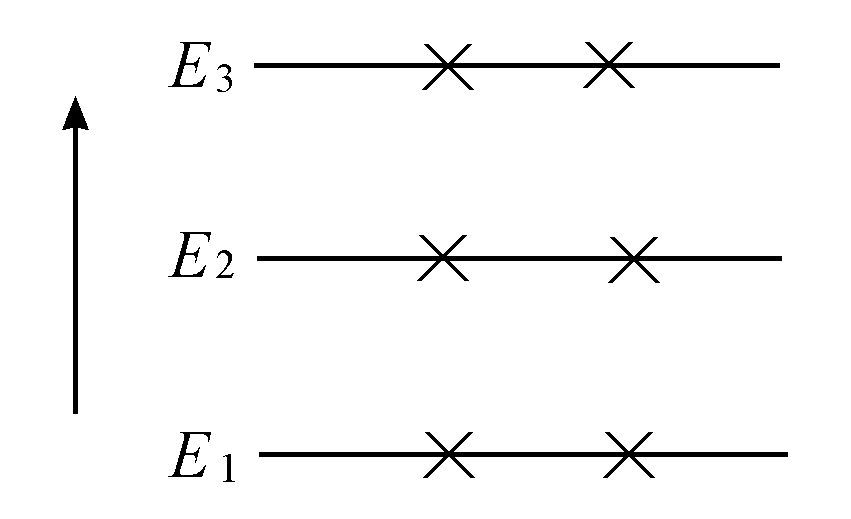
图4.2 高温分布
激发态与基态均被占据
玻耳兹曼的概率分布即方程4.6为我们提供了支配各种平衡态结构的基本原则。把它称做玻耳兹曼有序性原理可能是恰当的。它具有头等的重要性,因为它能描述多种多样的结构,例如包括像雪花晶体那样复杂、精致、美丽的结构(见图4.3)。

图4.3 典型的雪花晶体
玻耳兹曼有序性原理解释了平衡结构的存在,但还可以提出这样的问题:它们是我们周围所见到的唯一类型的结构吗?即使在经典物理学中,也有许多非平衡态导致有序的现象。当对两种不同气体的混合物加上一个热梯度时,我们就会观察到,该混合气体的一种成分将在热壁处增加起来,而另一种成分却在冷壁一端集中起来。这个现象在19世纪就已观察到了称为热扩散。定态中的熵,一般低于均匀结构中所应有的熵。这说明,非平衡态也许是有序的来源。正是这个观察,开拓了布鲁塞尔学派所创始的观点(有关历史概述见Prigogine,Glansdorff,1971)。
当我们转向生物学或社会现象时,不可逆过程的作用变得大为显著。即使在最简单的细胞中,新陈代谢的功能也包括几千个耦合的化学反应,并因此而需要一个精巧的机制来加以协调和控制。换句话说,我们需要极其精致的有功能的组织。还有,代谢反应需要特殊的催化剂——酶,酶是具有空间结构的大分子而且有机体必须会合成这些物质。催化剂是一种物质,它能加速一定的化学反应,但在反应中它自己并不被用掉。每种酶或催化剂完成一种特定的工作。如果我们看一下细胞所进行的复杂而有顺序的操作,我们就会发现其操作方式组织得简直就像现代的装配线一样(见图4.4,并参见Welch 1977)。
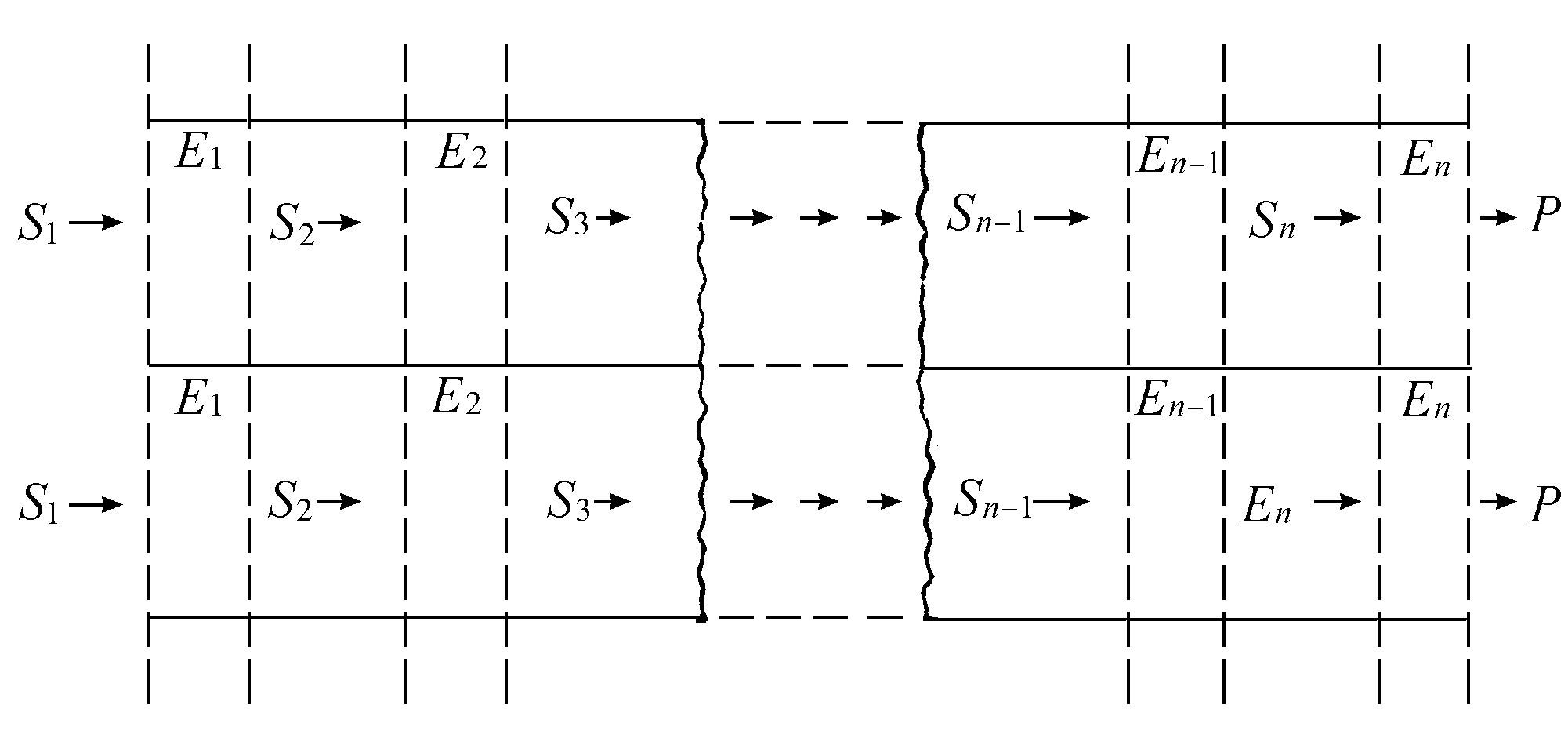
图4.4 多重酶反应的镶嵌模型
底物S1 通过一些连续变化在所“俘获”的酶的作用下变为产物P。
总的化学变化被分为一些连续的基元步骤,每步由一种特定的酶来催化。在图中,初始化合物用S1 表示;在每层膜当中有一种“被囚禁”的酶,对物质进行一定的操作,然后把它送到下一阶段。十分清楚,如此有组织的结构决不会是朝着分子无序方向演变的结果!生物学的有序性既是结构上的,也是功能上的,而且,在细胞的或超细胞的水平上,它是通过一系列不断增长复杂性和层次特点的结构和耦合功能表现出来的。这和孤立系统热力学所描述的演化概念正好相反,热力学的演化概念只是导致具有最大配容数的态,因而也就是导致“无序”。于是,我们是否必须像凯卢瓦(Callois,1976)所作的那样,得出“克劳修斯与达尔文不可能同时都对”的结论;或者我们应该与斯宾塞(Spencer,1870)一起,引入某个关于自然界的新原理,诸如“均匀的不稳定性”或“变异力——组织作用的创造者”等。
出乎意料的新特点是,如我们将在本章中看到的,非平衡态可以导出新型结构,即耗散结构(dissipative structures),它对于理解相干性和我们所居住的这个非平衡世界的组织作用,是很重要的。
线性非平衡态热力学
为了从平衡态转到非平衡态,我们必须用显函数的形式计算熵产生。我们不能再满足于简单的不等式,因为我们要使熵产生和确定的物理过程关联起来。现在,简单地求出熵产生已经成为可能,只要我们假定,即使在平衡态之外,也像在平衡态时一样,熵同样只与E,V,nr 等变量有关(在非均匀系统中,我们需假定熵密度依赖于能密度和区域浓度)。作为一个例子,让我们计算封闭系统中化学反应的熵产生。考虑如下反应:

在时间间隔dt中,由组分X的反应所引起的克分子数的变化,和由Y所引起的相等,而和A,B所引起的相反,即
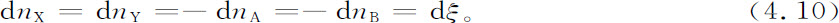
化学家通常在化学反应中引入一个整数νγ (正的或负的),叫做组分r的计量系数。ξ按定义是化学反应的进展度。于是我们可以写出:

反应速率是

考虑这个表达式以及吉布斯公式4.3′,我们立即得到

其中A是由德唐德尔(DeDonder,1936)首先引入的化学反应的亲和势,它和化学势μγ 有如下关系:

式4.13的第一项相当于熵流(见式4.1),而第二项相当于熵产生

利用定义4.12,我们发现单位时间内的熵产生取如下值得注意的形式:

这是不可逆过程(在此处是化学反应)的速率v和相应的力(在此处是A/T)的双线性形式。这类计算可加以推广:从吉布斯公式4.3′出发,得到

其中Jj 代表各种不可逆过程(如化学反应、热流、扩散等)的速率,而Xj 是相应的广义力(如亲和势、温度梯度、化学势等)。这就是宏观不可逆过程热力学的基本公式。
应该强调指出,为了导出关于熵产生的显函数形式4.17,我们使用了附加的假定。吉布斯公式4.3′的有效性只能建立在平衡态的某个邻域内,这个邻域规定了“局域”平衡的范围。
在热力学平衡态,对于一切不可逆过程,我们同时有

因此,至少在靠近平衡态时假定流和力之间有线性齐次关系,就是很自然的了。这种设想自然而然地包括了一些经验定律,例如说明热的流动与温度梯度成正比的傅里叶定律;表明扩散流与浓度梯度成正比的斐克扩散定律等。这样,我们得到下式所表达的不可逆过程的线性热力学:

不可逆过程的线性热力学受到两个重要结果的支配。第一个就是翁萨格所表达的倒易关系(Onsager,1931),这个倒易关系指出:

只要和不可逆过程i相应的流Ji 受到不可逆过程j的力Xj 的影响,那么,流Jj 也会通过同样的系数Lij 受到力Xi 的影响。
翁萨格倒易关系的重要意义在于它们的普适性。它们已受到许多实验的检验。它们的有效第一次表明了,非平衡态热力学也和平衡态热力学一样导出了与任何特定的分子模型无关的普适结果。倒易关系的发现确实成了热力学史上的一个转折点。
翁萨格关系的一个简单应用涉及到晶体中的导热性。按照倒易关系,不管晶体的对称性如何,热传导张量应是对称张量。这个重要性质实际上已由福格特(Voigt)在19世纪用实验的方法确立了,它相当于翁萨格关系的一个特殊情况。
翁萨格关系的证明可在一般教科书中找到(Prigogine,1967)。对于我们来说重要的是,翁萨格关系对应于与任何分子模型无关的普遍性质。正是这个特点使其成为热力学的一个成果。
应用翁萨格定理的另一个例子是两个容器所组成的系统,这两个容器用毛细管或薄膜连接起来,容器之间保持一定的温差。这个系统有两种力,比如说Xk 和Xm ,分别对应于两个容器间的温差和化学势差。其相应的流是Jk 和Jm 。当系统达到某个态时,其中的物质传输为零,而在不同温度下的两相之间的能量传输仍继续着,我们就说系统这时达到了一个非平衡定态。这样的态和平衡态之间不应有任何混淆,平衡态的特点是熵产生为零。
按照方程4.17,熵产生由

以及线性唯象定律(见式4.19)给出:

对于定态,物质流为零:

系数L11 ,L12 ,L21 ,L22 都是可测量的量,因此我们可以证明,确有

这个例子还能用以说明线性非平衡系统的第二个重要性质:最小熵产生定理(Prigogine,1945;亦见Glansdorff,Prigogine,1971)。很容易看到,方程4.23与4.24合在一起等价于下述条件:对于给定的常数Xk ,熵产生(式4.21)为最小。由方程4.21、4.22和4.24可得
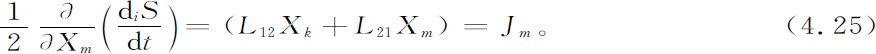
因此,物质流为零(式4.23)就等价于极值条件

最小熵产生定理表达了非平衡系统的一种“惯性”。当给定的边界条件阻止系统达到热力学平衡态(即零熵产生)时,系统就落入“最小耗散”的态。
建立这个定理的时候我们就清楚,这个特性仅在平衡态的邻域内才是严格有效的。多年来花费了巨大的努力想把这个定理推广,以便能适于远离平衡态的情况。但是令人深感诧异,最后证明的却是:在远离平衡态的系统中,热力学行为与用最小熵产生定理所预言的行为相比,可以颇为不同,甚至实际上完全相反。
值得注意的是,这种意外的行为在经典流体力学所研究的寻常情况中就已经观察到了。第一次用这个观点分析的例子是所谓“贝纳尔不稳定性”(Bénard instability)。(关于这个以及其他流体力学不稳定性的详细讨论,参见Chandrasekhav,1961)。
考虑一个在恒定重力场中两个无限平行板之间的水平液层,保持下边界的温度为T1 ,上边界的温度为T2 ,且T1 >T2 。对于“逆”梯度(T1 -T2 )/(T1 +T2 )的足够大的值,静止的状态变成不稳定状态,对流发生了。于是熵产生就增加,因为对流提供了热传输的一个新的机制(图4.5)。
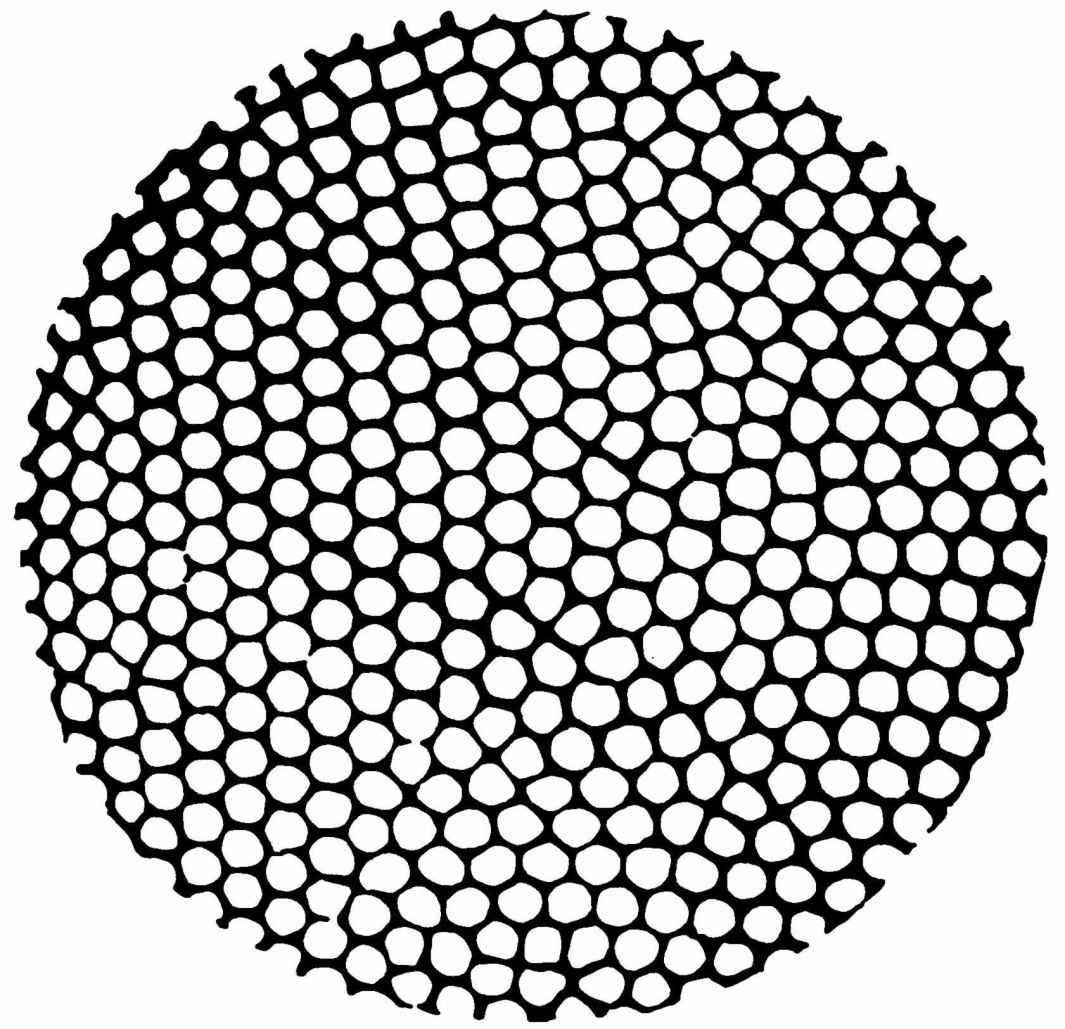
图4.5 在一个从下面加热的液体中,从上面看到的对流格子的空间花样。
此外,对流形成之后出现的流的运动和静止状态的微观流动相比是一个有高度组织的状态。实际上,为了得到一个可以辨认的流动花样,数目很多的分子要以相干方式在一个足够长的时间内移过可观察的距离。
这里,对于非平衡态可以是有序的起源这一事实,我们有了一个很好的例证。我们将在本章“化学反应中的应用”一节看到,不光是流体力学系统,对于化学系统只要满足加在动力学定律上的一些确定的条件,这一例证也确是成立的。
按照玻耳兹曼的有序性原理,出现贝纳尔对流的概率几乎为零,注意到这一点是很有趣的。只要新的相干态出现在远离平衡态的地方,包含在配容数计数之中的概率理论就不能应用了。在贝纳尔对流的情况中,我们可以想象总是有一些小的对流作为对平均状态的涨落而出现,但当温度梯度低于一定的临界值时,这些涨落将被阻尼并消失。若超过了这个临界值,则某些涨落被放大,并且出现宏观的流动。新的分子有序性出现了,它基本上相当于因与外界交换能量而稳定化了的巨型涨落。这个有序性的特点就是出现了我们所说的“耗散结构”。
在进一步讨论耗散结构出现的可能性之前,让我们简短地复习一下热力学稳定性理论的一些方面,这些将在有关耗散结构出现的条件方面给我们一些有益的知识。
热力学稳定性理论
热力学的平衡态,或者线性非平衡热力学中对应于最小熵产生的定态,都是能自动稳定的态。在第1章中我们已介绍了李雅普诺夫函数的概念。显然,在线性非平衡态热力学范围内的熵产生是这样的一个李雅普诺夫函数:如果一个系统被微扰,熵产生便将增加,但是系统则要发生反应以回到熵产生最小的态。对于远离平衡系统的讨论,再引入另一个李雅普诺夫函数是有用的。我们知道,孤立系统的平衡态,当对应于熵产生最大值时是稳定的。如果我们微扰一个接近平衡值Se 的系统,就得到

不过,因为在平衡态Se 时,熵函数S有最大值,上式的一阶项为零,所以稳定性由二阶项δ2 S的符号决定。
基础热力学的知识使我们能以显函数形式计算这个重要的表达式。首先考虑只有一个独立变量的微扰即式4.3′中能量E的微扰,则我们有
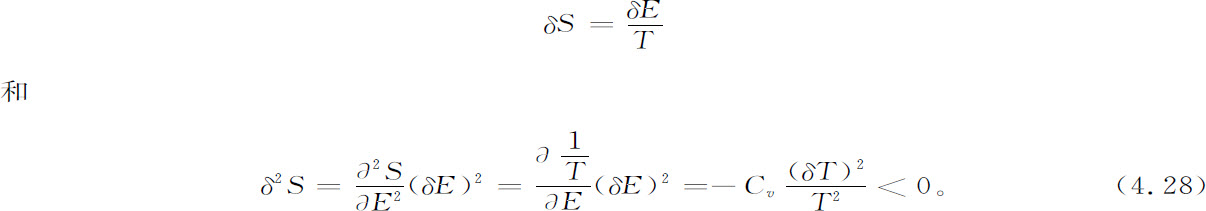
这里我们用到了这样一个事实,即定义比热为

并且比热是正的量。对更为一般的情况,如果我们对式4.3′中的所有变量都加上微扰,我们就得到一个二次型。这里我们只给出结果(计算过程可以在Glansdorff,Prigogine,1971的书里找到):
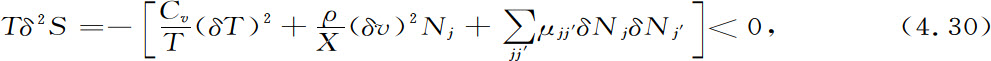
式中ρ是密度,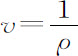 是比容(下标Nj 的意思是,当Nj 变化时,组分保持不变),X是等温压缩率,Nj 是组分j的克分子数,
是比容(下标Nj 的意思是,当Nj 变化时,组分保持不变),X是等温压缩率,Nj 是组分j的克分子数,
经典热力学的基本稳定条件是

这些条件的每一个都有简单的物理意义。例如,假若违反了条件4.32,则小的温度涨落不是被阻尼,而是通过傅里叶方程被放大。
满足这些条件时,δ2 S是一个负定的量。而且可以证明,δ2 S对时间的导数和熵产生P之间有如下的关系:
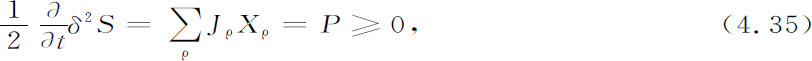
其中P的定义为

由不等式4.30和4.35得出δ2 S是一个李雅普诺夫函数,并且由于它的存在保证了对所有涨落的阻尼。这就是为什么在靠近平衡态时对于大系统有一个宏观描述就足够了的原因。涨落只起一个次要的作用,对于大系统来说,它们是定律的一个可以忽略的修正项。
这个稳定性能够外推到远离平衡态的系统吗?当我们考虑对平衡态有较大偏离但仍在宏观描述的框架之内时,δ2 S还起到李雅普诺夫函数的作用吗?为了回答这些问题,要计算微扰δ2 S,但现在是处于非平衡态的一个系统。在宏观描述的范围内,不等式4.30仍保持有效。然而δ2 S对时间的导数不再像式4.35中那样和总的熵产生有关,而是和这个微扰所引起的熵产生有关。换句话说,如格兰斯多夫和我(Glansdorff,Prigogine,1971)已经证明过的那样,我们现在有

右边就是我们所称谓的剩余熵产生。让我们再次强调,δJρ 和δXρ 是对定态Jρ 和Xρ 的偏离,而该定态的稳定性是我们正要通过微扰来检验的。和平衡态或近平衡态所发生的情况相反,方程4.37的右边(即剩余熵产生)在一般情况下具有不确定的符号。如果对于大于某固定时刻t0 (这里t0 可以是微扰的开始时刻)的所有t我们有

那么δ2 S就是李雅普诺夫函数,并且稳定性是可靠的。注意,在线性区,剩余熵产生和熵产生有相同的符号,我们就又得到最小熵产生定理的同一结果。但在远离平衡态的区域中,情况改变了。在那里,化学动力学的形式起着主要的作用。
下节中我们将举出几个化学动力学效应的例子。对于某些类型的化学动力学,系统可能变为不稳定的。这说明,在平衡态的定律与远离平衡态的定律之间有着本质的区别。平衡态的定律是普适的。但远离平衡态时,行为可能变得非常特殊。当然这是一个受欢迎的情况,因为它允许我们引入物理系统行为上的差别,而这种差别在平衡世界中是无法理解的。
假设我们考虑如下类型的化学反应:

其中{A}是一组初始反应物,{X}是一组中间产物,{F}是一组最终产物。化学反应方程通常是非线性的。因此对于中间浓度我们将得到许多解(图4.6)。在这些解当中,有一个对应于热力学平衡态,而且可以延长到非平衡区域,我们称之为热力学分支。重要的新特点是,这个热力学分支在离平衡态的某个临界距离处可以变得失稳。
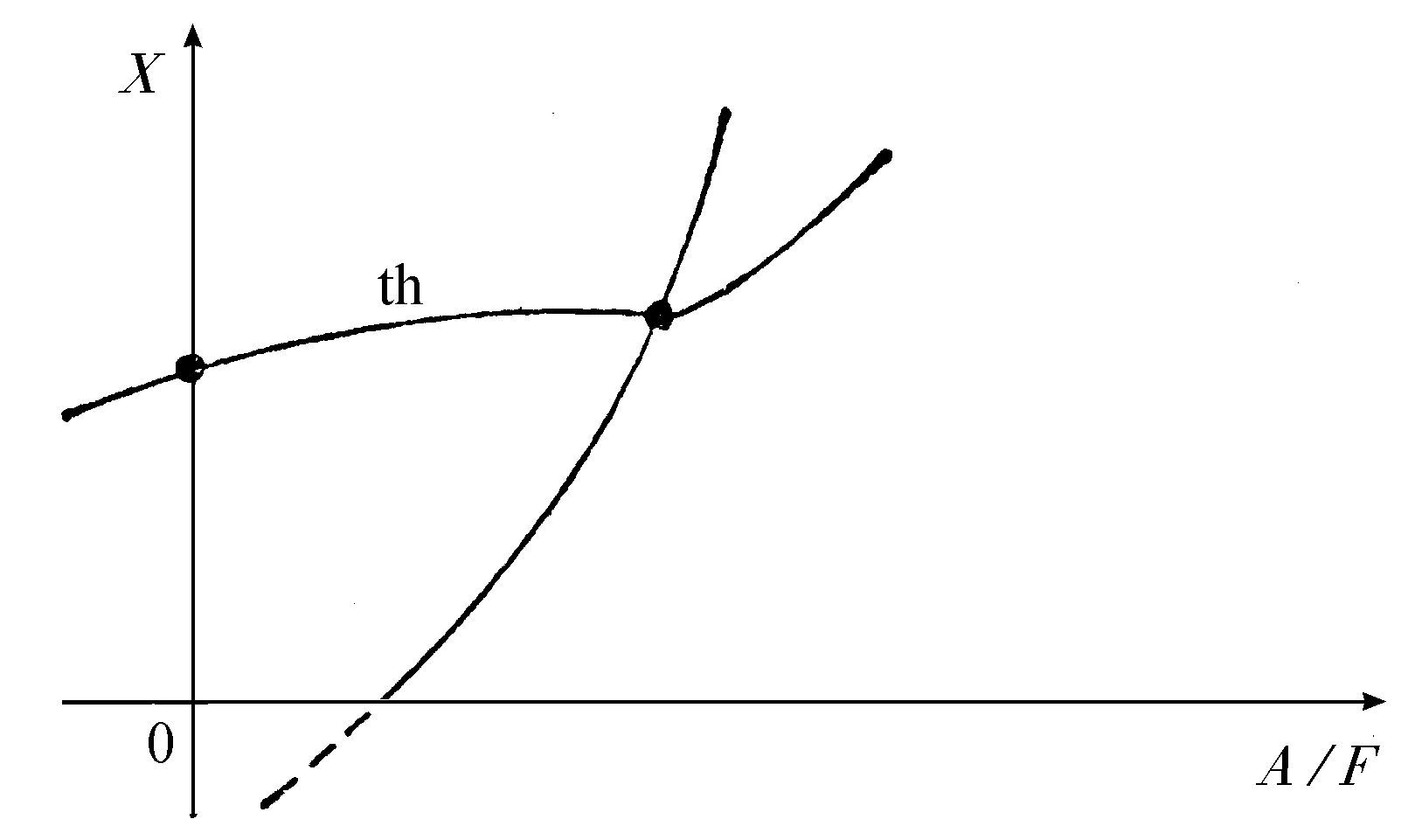
图4.6 对应于化学反应4.39的各种定态解
0对应于热力学平衡态;“th”是“热力学分支”。
化学反应中的应用
让我们把前面所述的形式体系应用到化学反应的情形。李雅普诺夫函数存在的条件4.38在这里变成

其中δvρ 是化学反应速率的微扰,δAρ 是方程4.14中所定义的化学亲和势的微扰。考虑下面的化学反应:

因为我们所感兴趣的主要是远离平衡态的情况,所以我们忽略逆反应,并对反应速率 (2) 写出

按照式4.4和式4.14,对于理想系统,亲和势是浓度的对数函数。因此,

浓度X在某个定态值附近的涨落,引起剩余熵产生;
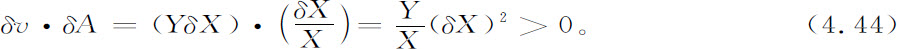
因此,这样的涨落不可能违反稳定条件(式4.40)。
我们现在考虑自催化反应(以代替式4.41):

仍假定反应速率由式4.42给出,但现在的亲和势是

现在我们得到对剩余熵产生的“危险的”贡献:
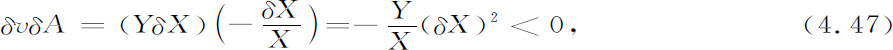
负号并非意味着微扰了的定态必然失稳,只是意味着有失稳的可能(正号是稳定的充分条件,但不是必要条件)。然而一般的结果是热力学分支的失稳必然引起自催化反应。
人们立刻想起这样的事实,即大多数生物学反应含有反馈机制。比如在第5章中我们将说明,对于活系统的新陈代谢来说是不可缺少的富能分子三磷酸腺甙(ATP),就是通过酵解循环中的一系列反应产生的;而这个循环在开始时就已经含有ATP了。为了生产ATP,我们需要ATP。另一个例子是,为制造细胞,我们必须从细胞开始。
因此,把生物系统中如此典型的结构同热力学分支稳定性的破坏互相关联起来是件非常诱人的事。结构和功能成为密切相关的了。
为了用一种清晰的方法掌握这个要点,让我们考虑催化反应的某些简单模式。例如

初始反应物A和最终产物E的值对于时间保持不变,所以只剩下两个独立变量X和Y。为简化起见,我们忽略逆反应。这是一个自催化反应的模式。X的浓度增长与X的浓度有关,Y也是一样。
这个模型广泛地应用于生态学模拟中,因为X可以代表使用A的草食动物,而Y可以代表以牺牲草食动物为代价而繁殖的肉食动物。这个模型在文献中是和洛特卡(Lotka)和沃尔特拉(Volterra)的名字连在一起的(May,1974)。
我们写出相应的动力学定律:

它们容许一个单一非零定态解:

为了研究在这种情况下和热力学稳定性相应的定态的稳定性,我们将要用正则模进行分析。我们写出:

以及

把方程式4.52代入动力学方程4.49和4.50,并略去x,y的高次项,我们就得到了对于ω的色散方程(色散方程表明齐次线性方程组的行列式为零)。因为我们这里有两个组分X和Y,所以色散方程是二次的,它们的显函数形式是

显然,稳定性和色散方程根的实部的符号有关。如果对于色散方程的每个解ωn 都有

则始态是稳定的。在洛特卡-沃尔特拉情况中,实部为零,我们得到
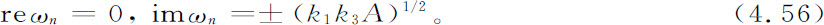
这就是说,我们得到所谓临界稳定。系统将围绕定态(式4.51)旋转。旋转的频率(式4.56)对应于小微扰的极限。振荡的频率和振幅有关,并且存在无穷多个围绕定态的周期轨道(图4.7)。
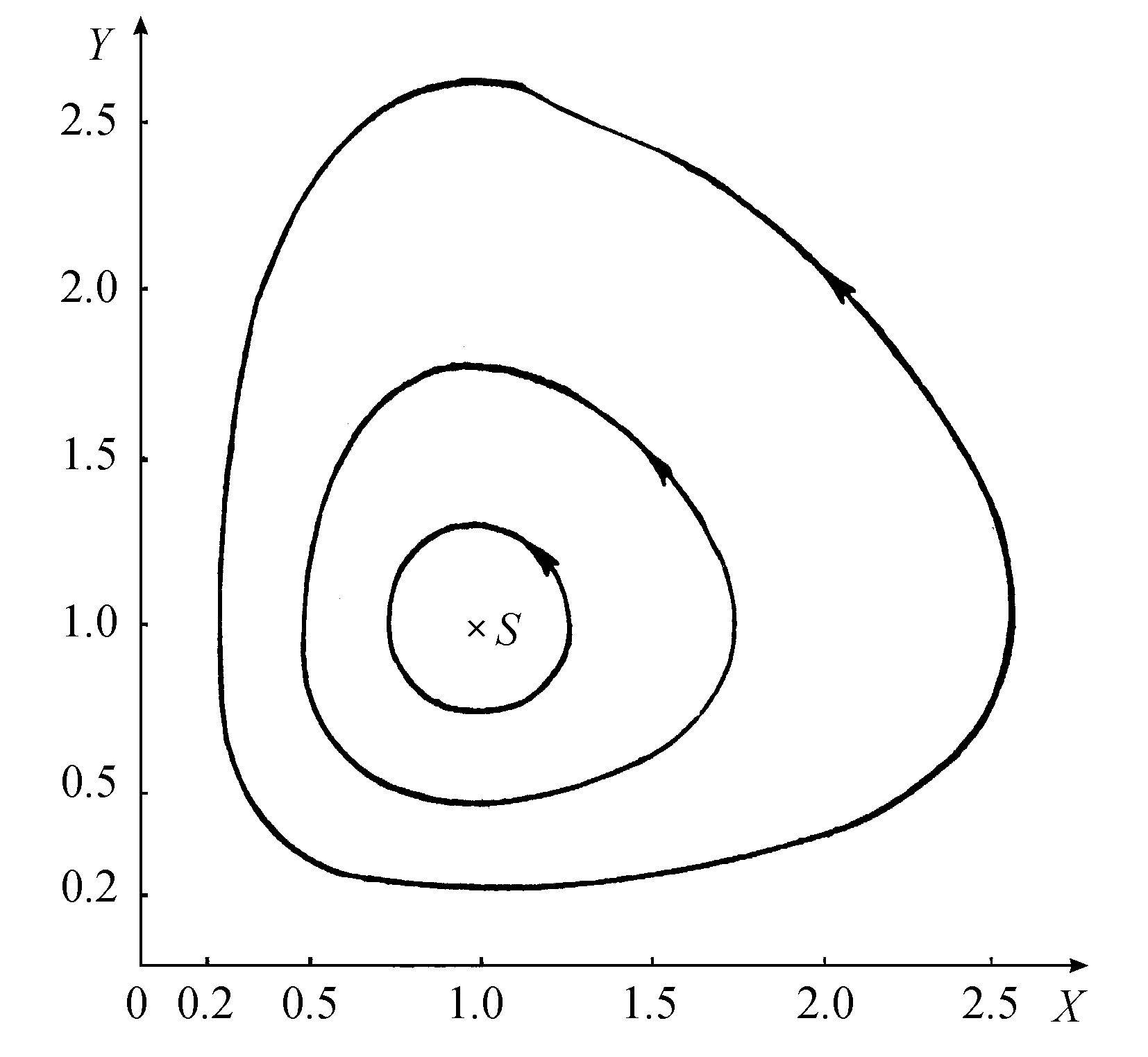
图4.7 对不同初始条件值所得到的洛特卡-沃尔特拉模型的周期解
现在我们考虑另一个例子,它近来已被广泛使用,因为它具有令人注目的特点,使我们能模拟广泛的宏观行为。这就是所谓布鲁塞尔器(brusselator),它对应于如下反应模式(详见Nicolis,Prigogine,1977):
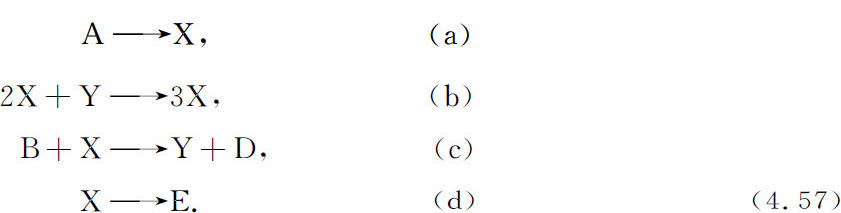
初始反应物和最终产物(A,B,C,D和E)仍保持不变,而两种中间组分(X和Y)可以有随时间变化的浓度。令动力学常数等于1,我们得到方程组:
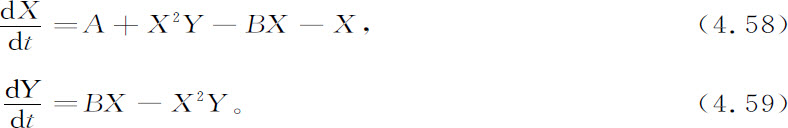
它们容许有下面的定态:

应用正则模分析,像在洛特卡-沃尔特拉的例子中一样,我们得到方程
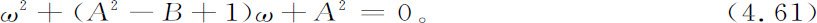
它应和方程4.54相对照。
我们立即发现,当

成立时,一个根的实部变为正数。因此,和洛特卡-沃尔特拉方程所发生的情况相反,这个反应模式给出一个真正的失稳。对大于临界值的B值所进行的数字计算以及分析工作得出了图4.8所示的行为。现在得到了一个极限环,就是说XY空间中的任何初始点都迟早要趋于同一周期轨道。重要的是要注意这个结果的非常意外的特点。现在振荡的频率变成了系统的物理化学态的一个十分确定的函数。而在洛特卡-沃尔特拉情况,我们已经看到,频率基本上是任意的(因为它和振幅有关)。
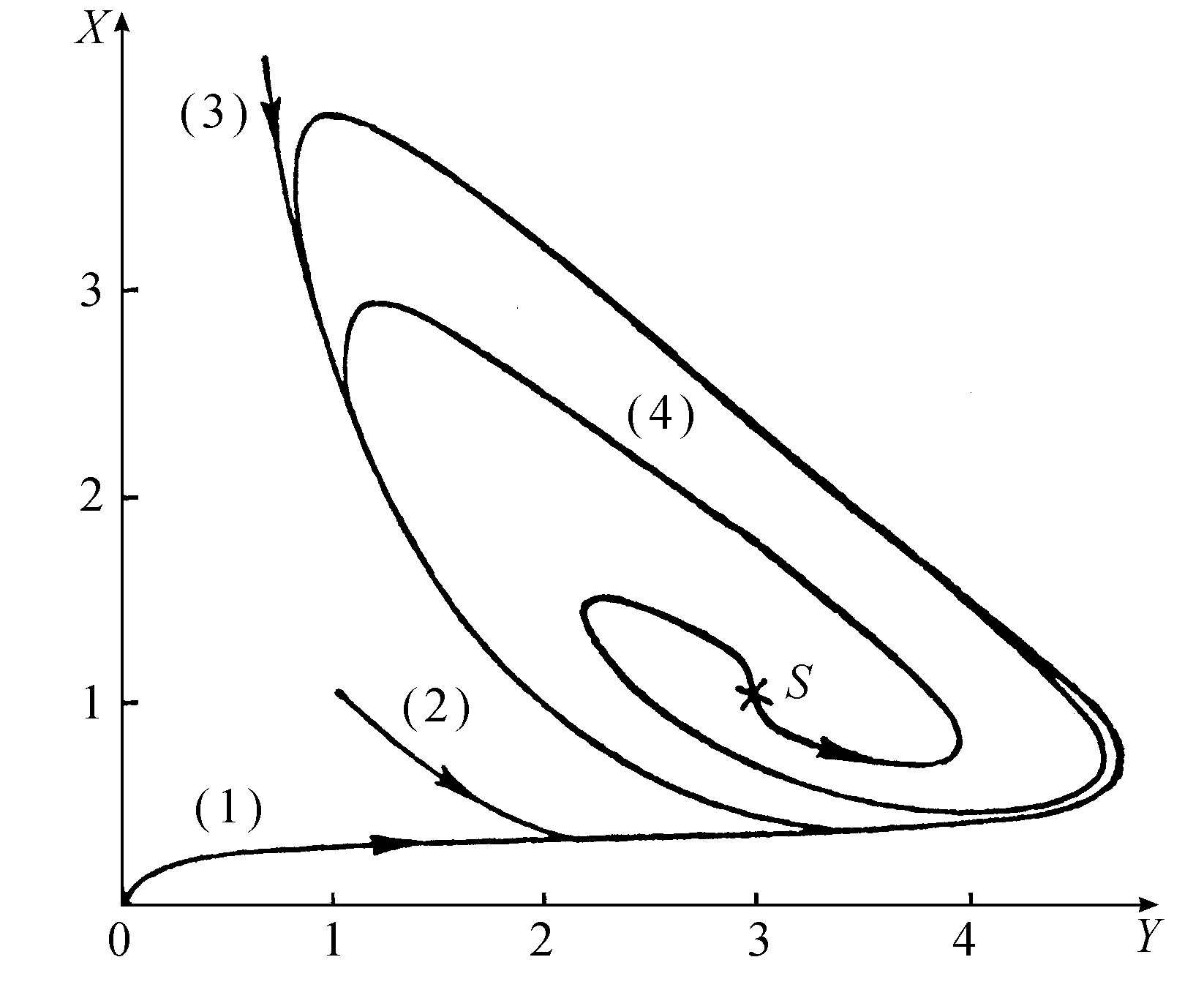
图4.8 布鲁塞尔器的极限环行为
不同的初始条件得出同一周期性轨道。S代表非稳的定态。
今天,已经知道了许多振荡系统的例子,尤其是在生物学系统中。而且重要的特点在于:一旦系统的态被给定,它们的振荡频率就是确定的。这说明,这些系统已超出热力学分支的稳定性。这类化学振荡就是所谓超临界现象。分子机制导出了十分引人而又困难的问题,我们将在第6章再回到这个问题上来。
极限环并不是超临界行为的唯一可能的形式。假设我们考虑两个盒子(盒1及盒2),它们之间有物质交换。代替方程4.58和4.59,我们得到方程组
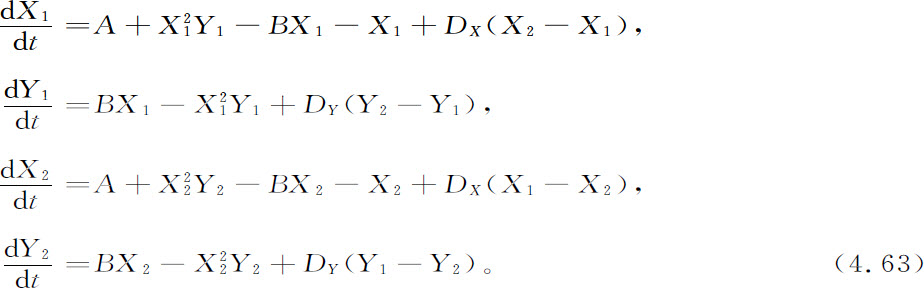
前两个方程是对盒1而言的,后两个是对盒2而言的。数字计算表明,在超过临界值的适当条件下,X和Y的相同浓度所对应的热力学态将变为不稳定的。X,Y的浓度由下式给出(见式4.60):

由计算机记录下来的这个行为的一个例子,示于图4.9。
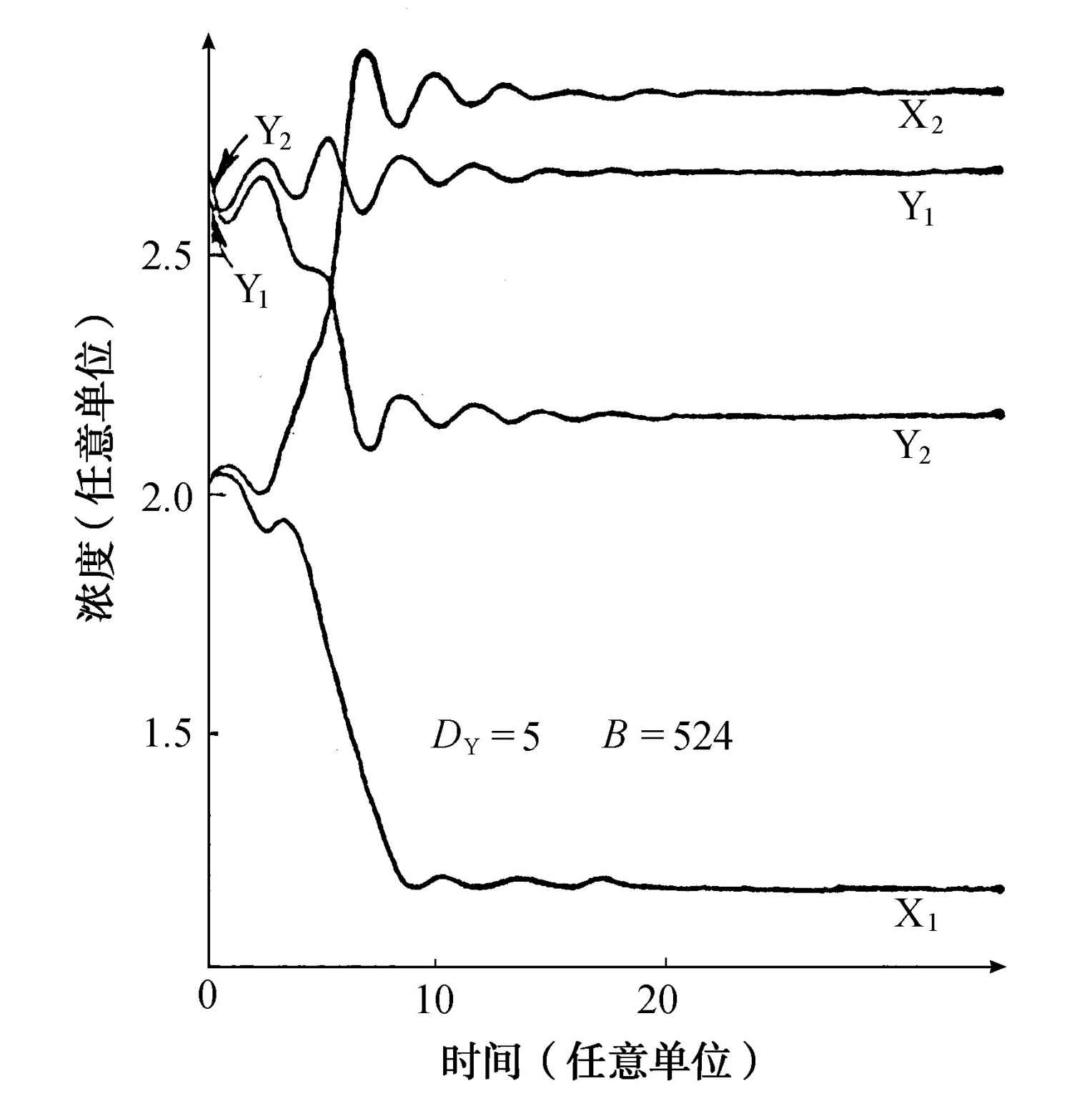
图4.9 盒2中的Y(即Y2 )围绕均匀态的微扰由于自催化的步骤而增加了在该盒中X(即X2 )的生成速率。这个效应不断增长,直到达到一个新的对应于空间对称破缺的态。
这里我们得到一个对称破缺的耗散结构。如果定态X1 >X2 是可能的,那么相应于X2 >X1 的一个对称的定态当然也是可能的。在宏观方程中无法说明将形成哪个态。
小涨落不再能改变组态,注意到这一点是很重要的。对称破缺系统一旦建立,就是稳定的。我们将在第5章讨论这些重要现象的数学理论。在结束本章的时候,我们要强调指出总是和耗散结构相连的三个方面:用化学方程所表达的功能;不稳定性所产生的时空结构;以及触发这个不稳定性的涨落。这三个方面之间的相互作用
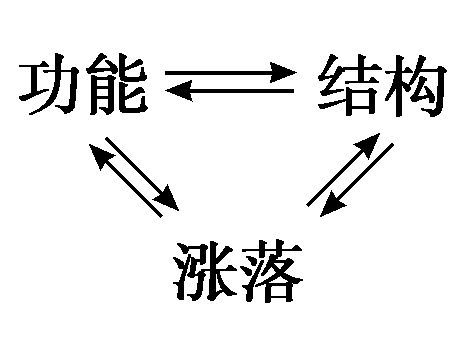
引出许多意想不到的现象,包括通过涨落达到有序,对此我们将在后面两章中加以分析。
————————————————————
(1) 理想系统的例子是稀溶液或理想气体。
(2) 为简化起见,我们取所有的动力学常数和平衡常数以及RT都等于单位1;并且把X的浓度CX 写作X等等。
